|
|
1. What is LOOPP?
2. How to interpret the results from the LOOPP
SERVER
3. Overview of LOOPP:
a)
from
the training set to recognition and back
b) deriving statistical potentials
c) overview of linear
programming training
d) pairwise and continuous pairwise
models
e) getting reduced representation
f) database files
4. Getting started
5. Fold recognition: quick start
6. Options and input files
7. LP training of a new potential
8. About the code.
9. Bugs and features.
10. References
|
by Jarek Meller and Ron Elber |
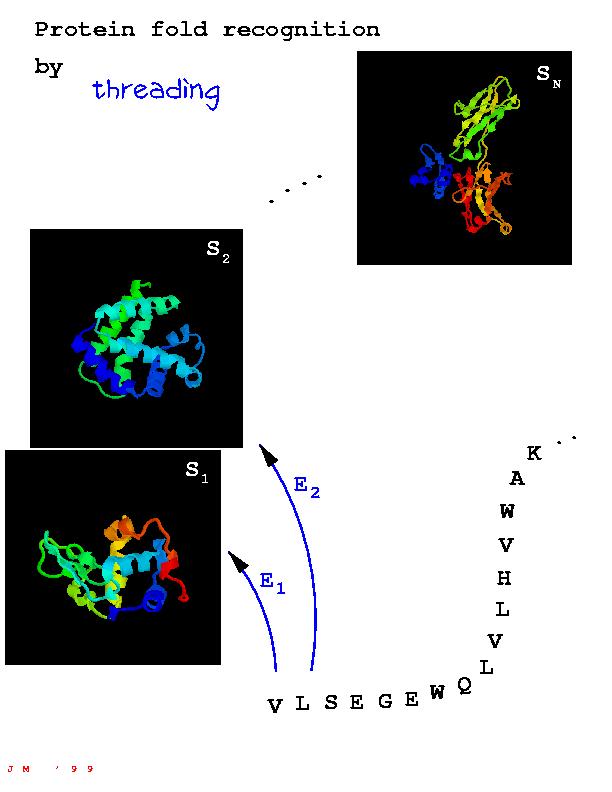
LOOPP is a program for PROTEIN RECOGNITION and design of PROTEIN FOLDING potentials. LOOPP (Learning, Observing and Outputting Protein Patterns) evolved from our previous LOOPP (Linear Optimization of Protein Potentials). As suggested by the new/old name there is some continuity, but there are also many new features.
LOOPP performs sequence to sequence, sequence to structure (threading), and structure to structure alignments. It further enables the optimization of potentials and scoring functions for the above mentioned applications. One may also use LOOPP to generate non-redundant libraries of folds, both for the training and recognition.
This page provides on-line help and a tutorial to "soften" the use of LOOPP. It also describes how to interpret the results of standard threading and sequence searches provided by the LOOPP server.
This section is meant to provide a brief description of the results from the LOOPP server. The LOOPP server runs the LOOPP program in the background. However, you are most likely simply interested in a plausible assignment of your sequence to a protein family (or structure and function), and not in all the features of LOOPP. If this is the case, you may safely ignore everything but the current section.
The results that you may have received by e-mail include the best global and local threading (sequence to structure) alignments, as well as (structurally biased) local sequence to sequence alignments. This is because the default search performed by the LOOPP server consists of three independent runs.
First, matching of the whole query sequence(s) into whole structures, included in the library of folds, is evaluated using global variant of the dynamic programming algorithm and developed by us (using LOOPP naturally!) THOM2 threading potential. Next, the same THOM2 potential and a local variant of the dynamic programming algorithm are used to evaluate compatibility between best matching fragments of the query sequence(s) and library structures. Finally, the best local sequence to sequence alignments are found using BLOSUM50 substitution matrix in conjunction with a gap penalty defined by a structural environment at a given site.
Our primary goal is to assign plausible structures to sequences that do not resemble significant sequence similarity to structurally (and functionally) chracterized proteins. To that end our primary predictions are based on threading and are complementary to standard sequence searches like BLAST or FASTA. However, we found out that our enhanced (structurally biased) sequence to sequence alignments are in some instances capable of detecting weak sequence similarities missed by PsiBLAST.
Besides, significant threading matches result from structures that are sufficiently similar. In some cases RMSD of only 5 Ang between the superimposed side chain centers may result in a weak signal. Of course, sequence based predictions are much less dependent on the variations in shape (as long as the sequence homology is preserved) and therefore including sequence alignments may correct for the effects of incomplete fold library.
Global threading alignments
Coming back to practical issues, let us analyse a sample of results
from the LOOPP server, using a cytochrome sequence (PDB code 1hrc) as a
query. The first section of the results should contain global threading
matches, ordered according to Z-scores, as printed in the third column.
First column contains names of the matches (library structures) using their
PDB codes with suffixes denoting chains. The second column contains energies
(as opposed to score, the lower the energy the higher the compatibility
between query sequence and library structure) and the last column contains
rough estimates of statistical significance (scale from very_low to VERY_HIGH):
|
The best matching structures (for query sequence 1HRC: CYTOCHROME_C) are: 5cytR ene=
-18.6 z_score= 4.3 ( 1.0)
significance= HIGH
|
Note that there are actually two different Z-scores printed in the third column. The primary one (equal to 4.3 in case of the best match) describes how the energy of a given alignment is related to the distribution of energies for random alignments. Z-score of 4.3 means that the energy of the alignment of 1hrc sequence into 5cytR structure is lower (i.e. better) by 4.3 standard deviations with respect to the average energy of a random alignment. Distribution of random alignments is obtained by shuffling a number of times the original query sequence 1hrc and aligning it to the structure 5cytR. The higher the Z-score the more significant is our match.
The second Z-score, which is printed in parenthesis and in case of the best match is equal to 1.0, describes how the energy of a given alignment is related to the native alignment. Using the same distribution of random alignments we find, in this particular case, that the energy of the alignment of 1hrc sequence into 5cytR structure is higher by 1.0 standard deviations than the native alignment of the 5cytR sequence into its own structure. In general, the closer the second Z-score to zero the better. See also THREADING.
Local threading alignments
Next, the results of the local threading search are included, using
similar format:
|
The best matching structures (for query sequence 1HRC: CYTOCHROME_C) are: 5cytR ene=
-30.5 z_score= 4.4 ( -4.3) significance=
HIGH
|
Significant threading matches are those with high global AND local
Z-scores. Our estimates of confidence
levels for the default threading searches used by the LOOPP server
are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, that in the above case we are left with only one significant hit, namely 5cytR. In fact, the example presented here nicely demonstrates difficulties that may occur in a truly blind prediction. Suppose that 5cytR is not included in the database, which is equivalent to a lack of sufficiently close 3D shape in the fold library. We still have two plausible matches: global into 1cdb and local into 1ccr. The first one is a false positive, whereas the latter one is correct. How can we know this?
A clue may come from the observation that 4 among the remaining 9 best local matches are cytochromes and also from the inspection of the alignments (see below). In general, however, we find assignments based on a single Z-score quite problematic since there are many false positives (e.g. 1cdb in this case) with relatively high Z-scores, overlapping with Z-scores of true positives.
Additionally, the default LOOPP server setup generates unconverged Z-scores that are computed for 100 randomly shuffled sequences. The actual Z-scores may be different by as much as 20% compared to the estimates provided by the default search. Therefore, we tend to believe only in those matches that are consistently resulting in high global and local Z-scores. However, if the energy of a global alignment is the lowest (and negative!) and its Z-score is greater than 4.5, then one may assign 90% confidence level to such match, even without significant local counterpart.
Local sequence alignments
Local sequence to sequence alignments are supposed to reveal matches into subdomains (unlike the threading searches, sequence alignments are NOT constraint to matches of length similar to the query sequence length). As such they may be less informative unless significant part of the query sequence is aligned to a database sequence. Therefore significance level printed to the right should be scaled down if less than 80% of the query sequence is aligned.
In the example presented here, however, there are no doubts whatsoever
- the whole cytochrome family is revealed with very high significance (note
that the distribution of Z-scores is energy function dependent and in case
of "diagonally dominated" sequence substitution matrix close matches result
in very high Z-scores):
|
The best matching sequences (for query sequence 1HRC: CYTOCHROME_C) are: 5cytR ene=
-590.0 z_score= 48.1 ( 10.2) significance=
VERY_HIGH
|
Threading predictions are seemingly more difficult for this family since, despite high sequence similarity, there is quite large variations in the corresponding 3D shapes. For example the RMSD distance between the superimposed side chain centers of 5cytR and 1ccr is equal to 6.9 Ang. When considering only the best local structure to structure alignment this distance decreases to 1.9 Ang. This is why 1ccr could be recognized by local threading alignment but not by the global one.
Threading vs sequence to sequence alignments
In light of the above, one might actually ask a question: do we need threading at all? The answer is YES (of course). In fact when sequence similarity is below detection level, even for the very best sequence methods as PsiBLAST, threading is our main (or perhaps the only) hope. Are you about to ask another nasty question - why LOOPP? Well, LOOPP employs novel threading potentials that uniquely blend profile and pairwise models, without the computational bottlenecks related to the latter ones. These potentials were trained using linear programming to recognize exactly large sets of protein structures and proved to be successful in detecting structurally related proteins that could not be identified with standard sequence methods. To find more, please read our paper or go to the bottom of this page.
Understanding the alignments
Coming back to practical issues once more, let us analyse the second
part of the results from the LOOPP server. It contains the actual alignments,
printed in the form exemplified below:
| query= 1HRC match= 1ccr energy=
-34.68 length= 96 type= local(seq-stru)
.......1.........2.........3.........4.........5.........6.
3 - 61
........7.........8.........9........
62 - 98
|
Although the sequence of 1ccr is not used while aligning 1hrc into the structure of 1ccr, the final alignment is printed using both sequences. The bars representing site numbers are also printed above and below (for query and match, respectively). Percentage of aligned parts of query and match are printed to the right. In this particular case 92.3% of 1hrc, from residue number 3 to 98, is aligned against 86.5% of 1ccr, from residue number 11 to 106, and sequence identity for the aligned parts is equal to 62.5%.
There may be quite many alignments for local searches, included in the
results. Local threading alignments are printed if the Z-score is greater
than 1.5 and local sequence alignments are printed if the Z-score is greater
than 2.0 irrespective of the rank. These alignments are not ordered and
may require FIND to navigate. It is however worth checking if a plausible
global match has a local counterpart with a Z-score higher than 1.5 (even
if it does not make it to the top). For example, the local alignment of
1hrc into 1cdb does not appear among the alignments that are printed, which
makes possible to conclude that 1cdb is most likely a false positve.
The prime OBJECTIVES of LOOPP are:
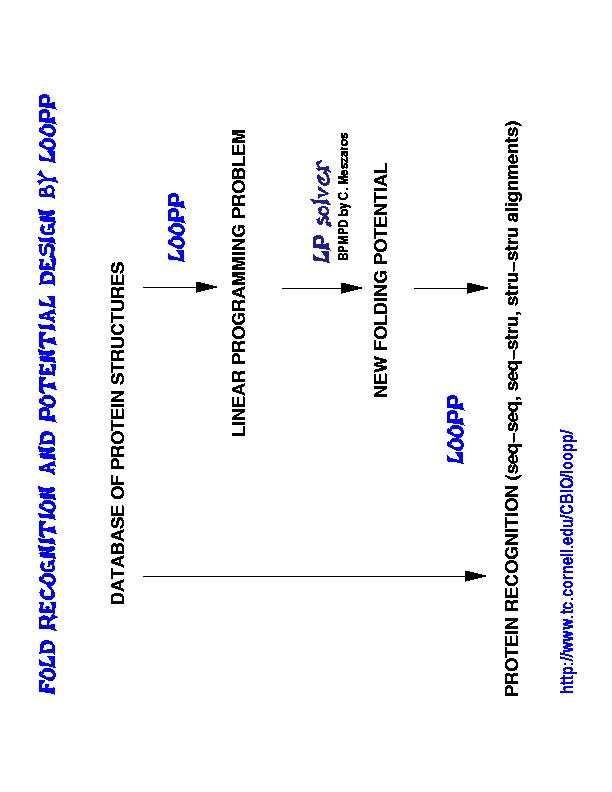
The interplay between design and training of folding potentials on one hand and protein fold recognition on the other hand is best depicted on the diagram included to the right (click on the picture to download enlarged version that can be printed).
Starting from a database of protein structures we may first employ LOOPP to develop various folding potentials (or scoring functions if you prefer) using the linear programming (LP) approach. These potentials are linear in their parameters and are trained by LP solvers in order to satisfy the requirement that the "energies" of the native structures are lower than alternative structures presented to the sequence.
Such trained potentials may be used for fold recognition. Query sequence is optimally aligned to library structures (or/and sequences) using dynamic programming and the best matches are picked up according to the lowest energy criterion (plus additional statistical verifications).
Finally, using novel structure to structure alignment method we may build non-redundant libraries of folds (of different sizes and similarity between representative structures) for further training and recognition.
B: Deriving statistical
potentials
Another possibility is to use LOOPP to develop statistical
potentials, based on the frequency of contacts or sites in a set of
representative structures. An extensive contacts/sites statistics may be
generated for most of the potential models.
C: Overview of linear programming training
In order to obtain the LP constraints gapless threading is performed (although explicitly generated decoys can be used as well) and the differences between native structures and the decoys (in terms of the functional model of the potential to be trained) are stored in files to be read by LP solvers. At present we use mostly the BPMPD program by Cs. Meszaros to solve the linear inequalities. However, other LP solvers (e.g. PCx, CPLEX) may be used as well. The optimized parameters define threading potential that recognize exactly all the native structures (against all the possible decoys generated by gapless threading) within the training set.
Alternatively, the problem may be infeasible i.e. there are no potential parameters that would make all the native energies lower than those of the decoys (within a given training set). This is clear sign that the functional form of the potential (or the content of the training set) must be adjusted e.g. one should increase the number of parameters.
D: Pairwise and continuous pairwise models in LOOPP
Some of the potentials that we develop aim at "ab initio" folding rather than threading. For example, continuous Lennard-Jones potentials that do not impose predefined cutoff distance are useful for the further structural refinement. Despite the NP-hardness of the problem of finding an optimal alignment with gaps, pairwise models are used in threading as well - either performing just gapless threading or employing the so-called frozen environment approximation.
E: Getting reduced representation
As an intermediate task the reduced representation of proteins (in the relevant formats) is generated. This allows to use LOOPP as a parser extracting the relevant protein characteristics for use with other programs as well. The general assumption is that protein structures are represented in terms of reduced models with one point per amino acid residue. These may be the positions of the alpha carbons or the centers of side chains or beta carbon positions. LOOPP can read and transform files in PDB and CRD formats into reduced representation. CRD format is used also by the molecular dynamics package MOIL. Thus, one can use MOIL to generate decoy structures for LP training of a folding potential.
The sequence query file can be in one of the following formats: FASTA, SWISS-PROT, plain 1-letter code, LOOPP compatible 3-letter code. The definition of the amino acid types (an alphabet) is very flexible and ranges from H/P two-letter alphabet to extended alphabets with user defined symbols.
F: Database (SEQ and XYZ) files
Coordinates of point residues of proteins belonging to some data set (training set or representative folds set) are necessary for the program to run. Since the allocation of memory is based on the content of the file with residue identities (sequences) this information is also required. Thus, irrespective of the choice of options (i.e. whether it is sequence to sequence alignment, which in principle does not need coordinate file, or whether it is sequence to structure alignment, which in principle does not need sequence file) both sequence and coordinate files (usually called SEQ and XYZ, respectively) are necessary to run the program.
The current version of the program has been used and tested on a couple of UNIX systems and should be easily portable to any UNIX platform. Besides, LOOPP was tested on Windows NT and Windows2000 (the LOOPP server runs on aWindows2000 PC). Although the source code is the same, there is an additional WinNT/2000 distribution file, which contains an appropiate executable and batch files instead of the UNIX shell scripts to run examples. Alternatively, you may just use the same UNIX distribution if you have Cygwin installed on your NT. LOOPP is written in C, there are no graphical interfaces (yet) and other fancy features, so in principle it should be portable to any other system as well.
In order to build the program you have to first uncompress and untar the distribution file. Type:
gunzip loopp2000.tar.gz
tar -xvf loopp2000.tar
The WinNT counterpart of gunzip and tar is winzip. The distribution file contains several subdirectories with the source code, documentation and examples. The source code is included in the subdirectory loopp/source. You may find a simple Makefile to compile it - type (in the source subdirectory):
make
(or nmake on NT). You should adjust Makefile to your local configuration if necessary. When the executable is successfully built it is moved to the subdirectory loopp/exe. To run it type (assuming that you are in loopp directory):
exe/loopp.exe -h
Option -h makes the program printing short help. There are several precompiled executables in the directory exe, so there are good chances that you do not need compilation at all.
To start using the program one needs basically three things:
a) file with sequences (with the default name SEQ)
b) file with coordinates of residues in reduced representation (XYZ)
c) file with query sequences for recognition (with the default name
seq_to_examine.txt)
Two databases are provided with the distribution, namely those developed by Hinds and Levitt and by Tobi and Elber. The latter one contains 594 structures and it is not fully representative. Larger (and more representative) library of folds are available from my own home page.
Suppose (optimistically) that everything went smoothly and loopp.exe
has been successfully created and it can be found in the CWD. Suppose also
that files containing library of folds (with standard names SEQ and XYZ,
where
SEQ contains sequences and XYZ coordinates of the protein residues
in the reduced represenation - point per amino acid) that are necessary
to run LOOPP are in the CWD too. Suppose also that we have a query sequence
of (say) unknown structure and we want to check which folds match best
our query sequence by the means of threading i.e. sequence to structure
alignment.
THOM2 is the default threading potential
THOM2 is right now the default model for the energy functional and our
currently best THOM2 threading potential
is included in the code as default. Therefore, we do NOT need to specify
a file with a potential, although it is of course possible to use another
potential with option -p.
Query sequence file and its format
We should type in our sequence - or sequences - (or paste it using the mouse) into a text file, that we may call for instance query.seq (or even better seq_to_examine.txt which is the default name that does not need to be specified explicitly later on). Depending on the format of the sequence we have to specify option -fmt (query format) to tell LOOPP how to read query.seq (format will NOT be recognized automatically and wrong format may lead to unexpected effects: most likely a message "Error while reading ... " would appear). The following formats can be used (note that both, lower and upper case letters can be used):
1. FASTA (-fmt fasta)
| >name_of_seq1
AAHHKLIaaGgTwAAGGDE >name_of_seq2 AgHHKLIaaGgEEYFFCAGGDE ... |
2. PDB (-fmt pdb)
The SEQRES part of the PDB file is read in this case (coordinates are only read if option -x for structure to structure alignments are used).
3. LOOPP (-fmt loopp , default)
The internal format of SEQ file is assumed:
| name_seq1 length_seq1
ALA ALA HIS HIS .... name_seq2 length_seq2 ALA GLY HIS HIS ... .... |
4. SWISS-PROT (-fmt swiss)
| ID 1433_ORYSA
.... ORIGIN 1 MSPAEASREE NVYMAKLAEQ .... ...... // ID .... |
5. Plain (or one-letter) (-fmt 1let or -fmt plain)
This format is used by the LOOPP server (use # symbol to separate sequences).
| aahhKLi ...
.... # aagHhiL ... ... |
loopp.exe -t query.seq -fmt <format>
where <format> is one of the defined above (for exmple fasta) and option -t stands for threading and corresponds to threading recognition loop. If we want to run threading with a non-default model (e.g. with a pairwise potential) we have to specify an additonal option:
loopp.exe -t query.seq -fmt fasta -eij
In such a case a default pairwise potential (of Dror and Elber) will be used. Since a pairwise model is employed only an approximate (first iteration of the frozen environment approximation) alignments will be generated. Frozen environment approximation is implemented inefficiently, since it was not the focus of our research. It is meant for checking individual alignments with a pairwise model (see option -pick).
By default global alignments are performed (as opposed to local ones). Twenty best, according to energy, alignments are subsequently subjected to statistical significance test. The so-called Z-score(s) are computed to measure the significance of a hit in terms of its distance from the expected score for a random sequence of the same length and composition.
By default 100 alignments for randomly shuffled sequences are performed for each putative match (structure) from the database (XYZ) to arrive at two Z-scores that are reported using stnd_out and are written into a file best.log.
Definition of the Z-scores
First (and the most relevant) Z-score, which is actually used to sort the best matches is defined as
- ( E_algn - < E_rand_algn > ) / sigma
and the second one (reported in the parenthesis) is defined as
- ( E_algn - E_nat_algn ) / sigma
where sigma is the standard deviation for the distribution of random alignments defined before. The bigger is the first Z-score the more significant is the match, on the contrary - the absolute value of the second Z-score should be small (if it is negative E_algn is better than E_nat_algn) for good matches. See also discussion of the LOOPP server results.
Local alignments as additional filter
Local alignments appear to be a valuble component of the quest for the
structure, since they provide an additional
consistency check. In other words it is much less likely that our match
is false positive (defined as incorrect match with good energy and high
Z-score, say larger than 3.0) if we consistently see the same match among
both, global and local hits. This is because the global alignments are
restricted to a subspace of alignmnets of a certain length and some Z-scores
may be still large just by the virtue of a similar length.
To run local alignments we need to add option -l (for local) to the command line e.g.
loopp.exe -t query.seq -fmt fasta -l
Modifying the default search depth
Unfortunately the energy itself is not a good filter for good local matches, so the Z-scores have to be computed for many structures. Therefore, to obtain meaningful results one needs to extend the search beyond the twenty (default for global threading) best structures, using option:
-best <n_best_to_be_included> <number_of_random_alignments>
For example one may type:
loopp.exe -t query.seq -fmt <format> -l -best 200 50
The number of alignments with randomly shuffled sequences is deliberately lowered to 50 to avoid too long calculations (50*200 alignments to be performed for each query sequence in this case). Once approximate Z-scores are calculated, one may converged the important ones using option:
-pick <query_name> <database_name>
For example to converge the Z-score for alignment of leghemoglobin sequence, which is called in our database 1lh2 (PDB-derived name) into a myoglobin structure 1mba we should type:
loopp.exe -t SEQ -l -pick 1lh 1mb -best 5 1000
Only the characters specified explicitly are compared which allows to
create somewhat more complicated
"picks". The -fmt option is not necessary since SEQ is assumed to be
in the LOOPP format (otherwise
program would not work properly anyway). One thousand random alignments
will be performed for each
of the 5 best matches with names starting with `1mb' - in this case
most likely the myoglobin structure
only (unless there are other structures with names starting with the
string 1mb).
Additional flexibility can be achieved using a keyword all for the pick option, as well as additional option -sd (for search depth), which may take values from zero to three (e.g. -sd 0; -sd 1; -sd 3). For example:
loopp.exe -t SEQ -pick all 1m -sd 0
will run global threading search for all sequences included in the Query file (taken to be just SEQ) against all the structures with names starting with the string 1m. Since option -sd 0 is used, however, structures with lengths that are different by more than 25% with respect to the query sequences will be excluded. Roughly speaking (the default) -sd 1 means only mild length-related restrictions (50% differences are accepted), whereas -sd 3 means no length-related restrictions.
Yet another variation on the theme is related to the option -list. One
may prepare a text file containing names of proteins to be included in
the threading loopp. If file my_list.txt contains:
| 1mba 1lh2 1pbx |
then typing:
loopp.exe -t SEQ -list my_list.txt
will result in threading all the sequences with names starting with either of the three strings into all the structures from the datbase that satisfy (the default) -sd 1 restrictions. Such list may also contain list of names of files in CRD or PDB formats, if additionally -fmt crd or -fmt pdb is specified.
Changing form of the output
The level of output is defined by option -i (level from one to four
i.e. -i 1; -i 2; -i 3; -i 4). The most useful one seems to be -i 1 (or
just -i) as it adds horizontally printed details of the alignments to the
file alignments.log
(see also best.log and xscan.log, as well as current.info
and protein.info, which are generated when -i 2 is used).
B: SEQUENCE TO SEQUENCE ALIGNMENTS
To perform the sequence alignment search (in which the query sequence
is aligned to all the sequences from
the database file - usually SEQ) we have to type a command similar
to the one used for threading:
loopp.exe -s query.seq -fmt <format>
When option -s (sequence to sequence alignment) is used a pairwise model is assumed and standard BLOSUM50 potential is employed. By default gap penalties are increasing with the number of neighbors to a given site. Thus, the default sequence alignments are structurally biased, becoming yet another mutation of mixed fold recognition methods. To perform "pure" sequence alignments with constant gap penalties one needs to add the option -cgap (for constant gap penalty). For example, assuming that query.seq is in plain (one-letter format) we may type:
loopp.exe - s query.seq -fmt 1let -cgap -g 0 8
to run so called overlap matches, which are global alignments without prefix/suffix penalties and with a constant gap penalty of 8 otherwise.
All the previous considerations about options -l ; -best; -pick; -i;
-sd; -list and many other not discussed here apply equally to sequence
alignment, which uses the same central engine and preserves the style of
output. As for the interpretations of the results, see: how
to interpret the results from the LOOPP server. Note also that in the
directory unix_shells one may find a unix shell to run combination of global
and local threading searches as well as local sequence search (called run_tgtlsl.sh),
which is similar to what the LOOPP server does.
C: STRUCTURE TO STRUCTURE ALIGNMENTS
Option -x is used to perform structure to structure alignments. All
the other options, discussed above, apply as well. The query file must
contain now not only the sequence, but also the structure. The formats
that are implemented are PDB (structure in PDB format against structures
from XYZ), CRD (structure in CHARMM
format against structures from XYZ) and LOOPP (which means that we
simply compare structures from XYZ file). Additionally, as for threading
and sequence alignment, the option -list can be used to specify a list
of PDB or CRD files, or a list of XYZ entries. The general form of a command
running structure to structure alignments is the same as for threading:
loopp.exe -x query.stru -fmt <format>
In order to perform standard structure to structure alignments for PDB structures included in the files 1MBO.pdb, 1HRC.pdb and 1A7K.pdb prepare first a text file (called say my_list.txt) containing paths to these three files and type:
loopp.exe -x -list my_list.txt -fmt pdb
D: BUILDING A NEW DATABASE
Building new (non-redundant) libraries of folds is closely related to structure to structure alignments. There are two basic operations that can be performed using option -x in conjunction with two other options, namely -newDB and -nored. Both of these option take as argumnet a name of the file, which is supposed to contain a list of non-redundant structures.
When using -newDB we actually assume that we want to extend an existing database, which is called "kernel". Suppose that our current XYZ file is such a kernel. In addition we may have a list of PDB (or CRD) files in a file all_pdb_files.txt. Typing
loopp.exe -x -list all_pdb_files.txt -fmt pdb -newDB
all the pairwise structure to structure alignments between the specified PDB structures and the database structures will be performed. At the same time a new list (in the default file new_list.txt) will be created will only these PDB structure names that are non-redundant i.e. they do not have any relative in the kernel that would be less distant than cutoff RMSD. The default RMSD cutoff is 3 Ang. In addition, 50% percent sequence identity threshold is used by default. These two parameters can be changed using options -trDB and -tsid, respectively.
The new list can be then used to create smaller addition to the kernel (with the redundant structures excluded). Next, using -nored we may check for redundancies within database (perhaps extended by new structures), typing:
loopp.exe -x SEQ -nored
Again, a new list (new_list.txt) will be created after performing all against all structure to structure alignmnets. This list will contain a subset of all the names of proteins included in SEQ. One may easily extract this subset from SEQ and XYZ using -list option. Type:
loopp.exe -x -list new_list.txt -justQ
where option -justQ makes the program stop right after creating new Query and Query_xyz file, which from now on become a new database. It is reasonable to change their names into something like SEQ_nored, XYZ_nored before they get destroyed by another run of LOOPP!
For large sets the procedure described above may be not quite feasible and it is better to divide the original database into parts and perform similar operations in an iterative fashion.
Model file and alphabet
Most of the options are read from the standard input, although the descriptions of the scoring function (threading potential) is defined in the file Model, which is sort of a primitive script. It is used to specify the following parameters: type of potential, number of parameters, alphabet, cutoff distances for non-continuous models. If file Model is found in the current directory, or if option -mod <model_file_name> is used to specify location of the Model file to be used, then it takes precedence over all the defaults. Thus, if pairwise model is specified in Model using eij keyword, then THOM2 (default potential) is no longer used. BEWARE of unexpected effects, if you use unintentionally Model file!
At present the order of parameters in Model is fixed:
| type n_par r_cut
ALA 10 ... |
where type may be eij (standard pairwise model), evdw (continuous pairwise model), ein (threading onion model (THOM) with first contact shell only - e_i(n) functional form), einm (THOM with first and second contact shell - e_i(n,m) functional form). After the first three parameters one may define an alphabet, which is different from generic one, giving explicitly symbols of letters to be used (the letters refer here to 3-letter codes of amino acids).
In the case of THOM models the number of types per letter is also read - for pairwise potentials it is obsolete but at present it has to be given anyway (one may put ones for all letters for instance). If an amino acid symbol is not specified in Model, then a replacement should be given, according to the chemical type of the amino acid. For instance when alanine is not specified, HYD is the expected replacement, otherwise the program stops. Similarly, if LYS is not specified, then one of the symbols: POL (for polar res.) or CHG (for charged res.) has to be given.
The list of non-standard symbols is the following (one-letter code in
parenthesis, short explanation to the right):
| GAP (-) - GAP
HYD (B) - hydrophoBic POL (O) - pOlar CHG (U) - plUs-minUs (charged - only positively charged if CHN used as well) CHN (X) - negative character (negatively charged) CST (J) - sulphur bridge (Joint CYSs) HST (Z) - attached ions with some Z e.g. Z=2 for Fe USR (=) - user defined |
The three last symbols (CST, HST, USR) do not imply any special treatment and there is no automatic conversion into it. These symbols are expected to be given explicitly in the sequence definitions of the file SEQ.
The generic alphabet and chemical types of residues are defined in functions
included in the file alphabet.c - there is no external file that would
allow changing the generic definition. The following residues are treated
as hydrophobic ones in first place:
| ALA, CYS, HIS, ILE, LEU, MET, PHE, PRO, TRP, TYR, VAL |
So, if you want to change the character of TYR residues, you need to give it explicitly in the definition of an actual alphabet in the file Model.
Ex1. In order to use potential e_i(n) with 12 different types
per residue Model should contain:
ein 240 6.4 ALA 12 ARG 12 ASN 12 ASP 12 CYS 12 GLN 12 GLU 12 GLY 12 HIS 12 ILE 12 LEU 12 LYS 12 MET 12 PHE 12 PRO 12 SER 12 THR 12 TRP 12 TYR 12 VAL 12 |
Ex2. In order to use e_vdw(i,j) model in turn, with 5 letter
alphabet (HYD,POL,CHG,CYS,GLY), Model should contain:
evdw 30 11.0 HYD 1 POL 1 CHG 1 CYS 1 GLY 1 |
The order of symbols in the specification of the alphabet is irrelevant. The ones after residue symbols are ignored, but they need to be given to make the program read the alphabet correctly.
Command line options
Regarding the command line options, first of all one may try option
-h to get small help. Besides there are some examples in directory examples.
Together with the examples included in this tutorial it should be enough
to figure out how to use the program. The list of options to be used in
the command line is the following:
|
-t <query_seq_file> threading -s <query_seq_file> sequence to sequence alignment -x <query_struct_file> structure to structure alignment -q generating ineq for LP training -d LP ineq for explicit decoys -i <#level> info and statistics, #level=1,2,3,4 -l local alignments -fmt <format> query seq (struct) file format that can be one of the following: fasta (f) FASTA swiss (s) SWISSPROT pdb (p) PROTEIN DATA BANK crd (c) CHARMM 1let (1) plain (simple one-letter code) loopp (l) LOOPP (default) -name <query_name> query name for query_fmt=1let -pick <query> <match> narrow the search to a given seq/stru -sd <#level> search depth, #level=0,1,2,3 -list <list_file> multiple query files specified in list -einm THOM2 potentials (default) -eij pairwise potentials -ein THOM1 potentials -evdw LJ potentials -p <pot_file> non-default potential -mod <model_file> non-default energy functional -best <#best> <#shuff> Z-score test range and convergence -nprn <#best> print only #best matches -g <#pre_g_pen> <#g_pen> gap penalties -cgap constant gap penalty (seq-seq) -cdel constant deletion penalty (threading) -nog use gapless threading for recognition -tl <#align_length_th> threshold for alignment length -tz <#z_sc_th> threshold for Z-score -tr <#rms_th> threshold for RMS distance -tsid <#seqid_th> threshold for sequence identity -te <#ene_th> threshold for energy -u take energies larger then ene_th -seq <seq_file> specify the sequence database file -xyz <coor_file> specify the coordinates database -newCM enforce recreating Contact Maps -k <#col> <#n_col> specify XYZ file format -o <out_file> specify the name of the stdout file -log <best> <aligns> specify names of the log files -mps <mps_file> write LP constraints in MPS format -w <dcon_file> write differences in contacts -frz <#n_frz> freeze (make constant) #n_frz first param -m read potential in matrix format -rsc <#scale> <neg> scale potential -pvdw <#rep> <#atr> LJ powers specification -b <#start> <#end> range of gapless threading in trainig -newDB <new_list> build (new) database -nored <new_list> remove redundancies from a database -trDB <#rms_th> RMS distance for exclusion from a database -justQ stop after building query files -dbg debugging -h help -v print version signature |
Database files
The sequences and structures of proteins are read from files SEQ
and XYZ and potential from
current.pot, respectively (if
not specified otherwise). The default format of XYZ file is (sc stands
for side chain coordinate):
| name num_of_res
x_ca_i y_ca_i z_ca_i x_sc_i y_sc_i z_sc_i ... |
Other possible formats are:
| name num_of_res
x_sc_i y_sc_i z_sc_i ... |
| name num_of_res
x_ca_i y_ca_i z_ca_i x_sc_i y_sc_i z_sc_i x_cb_i y_cb_i z_cb_i ... |
Use option -k to change the format and pick up right column. For example -k 1 2 means that the first 3-column block (the one corresponding to C alpha coordinates) in the first format - composed of two 3-column blocks - is chosen. If the third format - composed of three 3-column blocks - is used, then to read C alpha coordinates one needs option -k 1 3.
The default format of SEQ file is:
| name num_of_res
symbol_i ... |
where symbol_i denotes identity of the subsequent residues in terms of an alphabet defined by the user. To change names of the database files use options -seq and -xyz.
Potential file
The default format of the potential file is a vector of numbers in the order to be used by the energy function. In the case of pairwise models the upper triangle of an interaction matrix should be put row by row into a potential file.
Ex3. Suppose that we employ two symbols only and our Model file
contains:
| eij 3 6.4
HYD 1 POL 1 |
Since we specified symbol POL after symbol HYD in the Model, this order
will be assumed throughout the program. Thus, assuming that the interaction
matrix takes the form:
HYD POL HYD -1.0 0.0 POL 0.0 0.0 |
our potential file should contain three numbers, as follows:
| -1.0
0.0 0.0 |
One may also use potential file in the matrix form:
| -1.0 0.0
0.0 0.0 |
Option -m is required to employ matrix form. The order of the symbols and the number of parameters per symbol are read from the file Model.
Ex4. For example, when using e_i(n,m) model with two symbol alphabet,
Model should contain:
| einm 30 6.4
HYD 15 POL 15 |
and potential file should contain 30 numbers, first 15 corresponding to hydrophobic residues and then 15 corresponding to polar residues.
Coarse graining in THOM2
There is fixed coarse-graining in the current version of the THOM potential
with two solvation shells (einm), which we found sufficient for protein
family recognition. The number of neighbors is defined with a cutoff distance
specified in Model. Each site has specific number of neighbors, such that
it belongs to one of five categories:
| 1. with very small number of neighbors (up to two),
2. small number of neighb. (three or four), 3. medium number (five or six), 4. large number (seven or eight), 5. very large number of neighbors (nine and more). |
Then, each neighbor may have different number of its own neighbors,
such that it belongs to one of three categories:
| 1. with small number of neighbors (up to two),
2. medium (between three and six), 3. large number of neighbors (seven and more). |
In this way each contact is characterized by two numbers n and m, defining the categories of the two sites involved in the contact. While threading we consider only the identity of site (i) that we are currently occupying. The result is the e_i(n,m) contribution for a contact involving site i of n neighbors with another site (of unspecified identity) of m neighbors.
Thus, the 30 parameters in the potential file may be logically divided into five consecutive blocks with three numbers in each. In the potential file we would simply have 30 numbers, separated by at least one space character. Using option -i 2 (info) makes it possible to check whether the order of parameters in the potential file is correct. With this option the program prints the potential that is actually used (into the file current.info) in a format that is easy to understand.
Generating statistical patterns with option -i
With -i (-i 1 to -i 4) the program prints some other useful information:
1. contact and site statistics, the contributions of all the parameters
to the energies of the native structures in the data set used (whole data
set information),
2. for pairwise models a statistical potential resulting from the frequencies
of contacts in the database is derived and printed to the file current.info
3. contact and site statistics, information for particular proteins
(stored in file proteins.info),
4. comparison of statistics of contacts and sites for native vs decoy
structures,
5. analysis of effective pairwise contributions for THOM2 model,
6. histogram for energy differences (between native structures and
decoys) distribution.
Certainly, it would be nice to have more advanced system with a graphical interface that would allow to prepare input easier, but this has to wait. Well, science is more interesting than just programming ...
In the learning phase, linear constraints are generated to allow optimizing a new potential by LP solver (not provided in LOOPP!). This mode of operation requires option -q. The threading is performed for sequences of known structures, comparing native energies with the energies of the decoy structures, to compute the linear inequalities. All the possible gapless alignments are generated. Each alignment generates one inequality.
Another possibility is to put explicitly generated decoys into XYZ and SEQ files and use option -d to generate inequalities with respect to the native structure, which is assumed to be the first one in XYZ and SEQ files.
Energy differences between the native and decoy structures are printed to a file xscan.log whereas differences in contacts are put onto a file current.dcon (if -w is used) or current.mps (if -mps is used). It is actually better to communicate with the LP solvers without the intermediate step, in which the MPS format file is generated by loopp and then read by the solver. MPS format is outdated and it implies usage of large memory and disk resources, especially when the primal problem is generated. The best strategy is to generate the dual problem in a compressed format understandable for the LP solver. Therefore option -w which generates just the coefficients of the constraint matrix in row fashion is more handy.
In the subdirectory loopp/examples/lp_training; test and explicit decoys you may find a number of potentials from the literature or potentials developed by us, as well as small data sets of 31 proteins. In the examples described below we will make use of them to demonstrate various features of the program that are related to linear programming training of new folding potentials.
There is one new feature, which is not discussed here in detail, namely the option -nfrz which allows to freeze a number of first n_frz parameters. For the frozen parameters, their contribution is simply calculated using current guess and it is moved to the right hand side of inequalities (additional file is created for that purpose). The remaining free parameters can be then optimized. This option has been used to train gap penalties with the rest of the parameters kept unchanged.
Ex7a. Developing a new potential
In our first example we will assume that we want to find a standard pairwise potential solving given set of proteins i.e. we want to get 210 parameters, such that the native energies would be always lower than the energies of decoy structures generated by means of the gapless threading of the native sequence through all the structures in the data set.
There are many pairwise potentials, that have been developed by now, and one may use one of them to start with (although it is not necessary - we may just assume that our initial potential is random). We picked up Hinds-Levitt potential. If one performs (gapless) threading loop to compare energies of decoys with those of native structures, one finds (see next example) that there are three structures with energies higher than some decoys, according to Hinds-Levitt potential. Example in the directory learn shows how to use LOOPP to generate inequalities to be solved by an LP program in order to get a potential, that correctly identifies all the proteins in the set.
Assuming that the files XYZ and SEQ are in the current directory, the command (as used in shell script learn/run_ex.sh) includes the option -q which generates the LP constraints:
loopp -q -p hinds-levitt_eij.mat -m -te -w hl.dcon
will produce the file hl.dcon with the coefficients of the constraint matrix. It then may be read by an LP solver providing new solution in parameter space or a confirmation of the infeasiblity of the problem. Alternatively, one may use the option -mps to generate the LP constraints in the MPS format.
Running run_ex.sh in directory learn generates both hl.dcon and hl.mps files, which are then compared with their templates from directory output_tmpl. If everything is fine the file out_diff should be empty after completion of run_ex.sh (simply type run_ex.sh to run it).
Option -m in the above example means that we want to use the potential in a matrix form, whereas -t means that we want to generate only those inequalities that are still not satisfied, given current potential. One may specify threshold for energy differences e.g. -te 10 to get only those with energy differences (with respect to native structures) smaller than 10 (it is not always feasible to generate all the inequalities).
Ex7b. Testing new potential
Let us now assume that we already developed some potentials and we want to test their prediction capacity. Before we do a real test, however, trying to recognize an unknown structure, we may check our potentials with known structures, which were not included in the training set.
In directory test we provide a realistic example of this type, with two potentials developed using Hinds-Levitt data set: one by Hinds and Levitt themselves (employed already in the previous example) and one developed by us using LP protocol and LOOPP. The potential me_r10_eij.pot is defined in terms of a reduced alphabet (consisting of 10 symbols only - see file Model_r10_eij) and it provides a nice demonstration of this additional flexibility of loopp.
Command lines from the script run_ex.sh in this directory:
loopp -q -p hl_eij.mat -m -te -o hl.log
loopp -q -p me_r10_eij.pot -te -o me.log
generate two log files (hl.log and me.log) that first of all should
be compared again with templates in the directory output_tmpl. Their content
is worth a closer look. An example of a line from a log file is given below:
| 971 -2.250000 1ptx 1bbt1:66 129 |
First, the number of a decoy (or gapless alignment) is listed, then its energy with respect to native one. In the next two columns we learn that it referred to the alignment of sequence of the protein 1ptx (PDB codes are used for names of the proteins) against structure of the protein 1bbt1, starting from residue 66 and ending at residue 129 in the 1bbt1 structure. Apparently, the other alignments of 1ptx against 1bbt1 (e.g. 65 128) had energies higher than the alignment of the sequence of 1ptx against its own structure (i.e. native energy) and they were not printed as we used -t option with the default threshold equal to zero.
It is interesting to note that although the number of inequalities,
which are not satisfied is larger in case of our me_r10_eij.pot potential,
the number of proteins that were not recognized is smaller. It is equal
to two, whereas in case of Hinds-Levitt potential (which employs full alphabet
and 210 parameters instead of 55 parameters of the reduced model)
there are three proteins having decoys better than native structures.
Ex7c. Training
a potential using explicit decoys
Suppose now that you generated a number of decoy structures performing MD or Monte Carlo simulations. Thus we have the native structure and a collection of perturbed structures and we want to train a potential that would distinguish native structure with respect to all perturbed ones. First of all one needs to translate full atomic representations (if it was used in MD/MC run) into reduced representations. If you used MOIL, then LOOPP (together with the script run_crd2ca-sc.sh or run_ex.sh in the directory red_repr) may help you - otherwise you need to use your own interface.
If the decoy structures are already translated into reduced representation, then we may use option -d to generate LP constraints for them (in conjunction with option -w or -mps) or to scan all the inequalities to check the energies of alignments with respect to the native energy. There is one thing to keep in mind - the native structure is assumed to be the first one in the data base that we read.
Thus, if all the structures are in the file XYZ_1bpt and all the sequences (which are in this case identical) in the file SEQ_1bpt, we may type:
loopp -p evdw_1bpt.pot -pvdw 12 6 -seq SEQ_1bpt -xyz XYZ_1bpt -d -k 1 1
to get the energies of alignments of the native sequence against all the decoys (with respect to the native energy) in the file xscan.log.
To illustrate simultaneously some other features, we use here a continuous
pairwise model of the potential (option evdw with the Lennard-Jones powers
12 and 6 : option -pvdw) and moreover, we use alpha carbons coordinates
in the reduced representation. They are assumed to be given in the first
(and the only) three column block of the XYZ_1bpt file and therefore
we need to use option -k 1 1 in order to choose the right format of the
coordinate file.
This is certainly a code in statu nascendi, so I cannot give any warranty. Here is a couple of hints about the organization of the code.
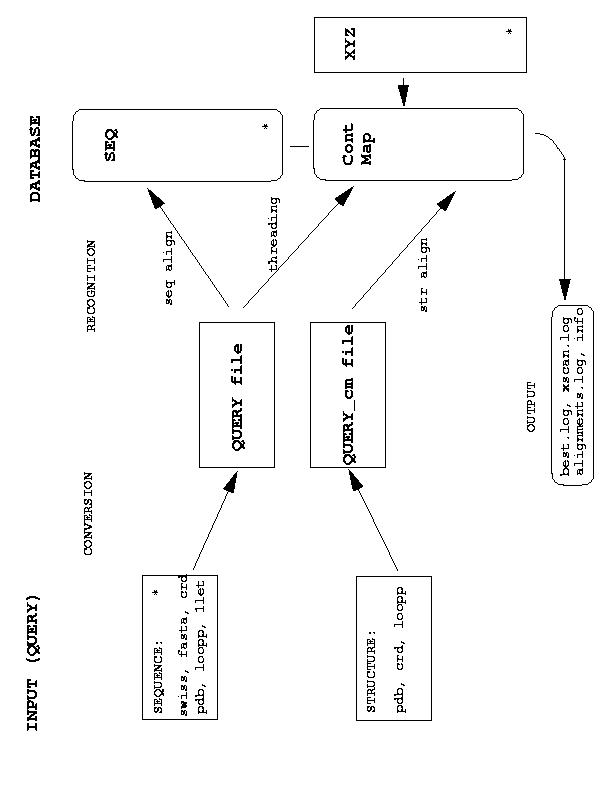
Functions that translate different query formats into internal Query files are included in the interfaces.c and they are called from build_irreps() function (see irreps.c). The actual sequence, threading and structure alignments are performed in align.c, thread.c and stru2stru.c, respectively. However, they use the same bunch of functions for dynamic programming, as included in align.c.
Arrays used in the program to store data are allocated according to what is found in Model file and according to the content of SEQ file, which allows to define max_length, max _prot and other parameters used in function alloc_mem(). The allowed length of strings (and thus of the file names) is given by MAXS - if it is too short you need to adjust MAXS and recompile.
Variables with names containing one capital letter represent (in most cases) arrays (including strings). There are very few multiple index arrays in the code, so in most cases arrays are just vectors. The pointer-offset notation for arrays is used throughout the code (with few exceptions again). Since the code still evolves rapidly there is no protection of the variables when they are used within functions. In many cases they are not supposed to be modified and they should be declared const to avoid accidental overwriting.
As for the options, they are kept in the structure keep_options (see the definition file loopp.h). The options eij, ein, einm, evdw, r_cont are treated in a special way and are specified in the Model file. All the others are to be specified in the command line (see examples). The default values of options are defined in function set_default_opt(). The default names of all the files specified in the function set_file_names(). All the names are chosen such that they should be self-explanatory.
Files ContMap, ContMap_vdw_A, ContMap_vdw_B are used to store pointers to neighbors of each site in each structure belonging to a data set. They are recreated, if not found in the current directory or if found inconsistent with the current database. File cont_by_Type is recreated every time and is used to store protein representations in terms of contacts. This precomputed representation is then used to make threading loops efficiently. The issue of efficiency was nevertheless somewhat less important, so there is a lot of room for improvements.
Each option must be given independently, with the hyphen sign preceding it i.e. notation -ih cannot be used as a shortcut for -i -h
Floating numbers are used throughout the program and certainly the accuracy of numerical operations is limited. It may cause some small discrepancies in terms of energies while comparing new results with the templates provided in examples directory. It should concern, however, only the last digit - if you see more than that, something may be wrong.
The option -i (info), which produces more complete output and generates statistics, is not fully implemented for the continuous pairwise models of the Lennard-Jones type (option evdw). The option -mps will not work with evdw either.
While rescaling the scoring (or potential function) using option -rsc one cannot give negative value directly, but rather use additional string to indicate that minus sign should be taken e.g. -rsc 1 neg to multiply the scoring function by -1. The same concerns option -t while specifying the selection threshold for the storing of inequalities.
The alignments are implemented assuming that we deal with an energy rather than a scoring function and will not work properly unless you use option -rsc to define properly rewards and penalties. Alignments assume also that the border between rewards and penalties is zero (as in the standard Smith- Waterman algorithm) and will not work properly with a scoring matrix that is e.g. all positive.
The THOM model with the second solvation shell (option einm) will work properly only when the number of parameters per residue is equal to 15. This is a result of prefixed coarse-graining that may be changed only on the source code level at present.
A dangerous situation may happen when you change for instance cutoff distance in the Model - program will work using previously generated ContMap (since it cannot recognize that cutoff distance has been changed and ContMap is no longer appropiate). The results, although looking reasonably will be wrong. Remove file ContMap to get it rebuilt whenever you change cutoff distance (or use option -newCM to enforce recreating ContMap file).
Beware of the content of the file Model - if you use THOM-like definition
for an option requiring pairwise model (e.g. sequence alignment with option
-a) or pairwise model for something requiring THOM-like model (e.g. structure
alignment), then you may get wrong results without warning.
Please cite:
J. Meller and R.Elber; "The
design of an efficient and accurate threading algorithm: Choice of energies
and statistical verifications", submitted
As it may be used as a general introduction to our approach to the threading
problem, the abstract of the talk that JM presented at the International
Conference on Optimization in Computational Chemistry and Molecular Biology
at Princeton (May 1999) is included below:
Novel folding potentials for efficient threading based on optimization with linear constraints.
The threading problem is known to be NP hard [1], when standard pairwise potentials are used. In this abstract a different kind of potential is used to solve the problem in polynomial time. The proposed computational study relies on local energy functions in the spirit of the work of Eisenberg and his collaborators [2] and on linear optimization of the potential energy parameters [3]. To speed up conformational searches and evaluation of energy function proteins are frequently represented at the residue level (reduced representation). Two widely used classes of reduced folding potentials are based either on pairwise interactions [4-8] or on the local structural environment (profile) of the amino acid [2], respectively. We are using profile energy functions of our own design. The local energy function has the advantage that the threading (with gap) can be performed in polynomial time, due to the use of dynamic programming. Given a functional form of the potential, the energies of all the decoy structures are required to be larger than the energies of the native conformation. If the potential is linear in its parameters, as is the case in numerous formulations, the parameters can be found efficiently with the LP approach. We trained a set of 594 representative protein sequences and structures [8] using gapless threading. A total of 3*10^7 inequalities are solved. We consider a class of `solvation like' potentials with energy that depends only on the identity of the site residue and the number of its neighbors in the first and second solvation shell. We call it THOM (THreading Onion Model). The model is local, and thus enables efficient threading search, and it is demonstrated to have recognition capacity comparable to that of pairwise potentials. The parameters of the potential can be improved in a systematic way by imposing more constraints on parameter space. Further developments in the functional form may follow. References: 1. Lathrop RH; Protein Eng 1994 Sep;7(9):1059-1068
|
LOOPP has been used to identify a novel plant gene, which controlls the size of the tomato fruit. See:
A. Frary, T.C. Nesbitt, A. Frary, S. Grandillo, E. van der Knaap, B.
Cong, J. Liu, J. Meller, R. Elber, K.B. Alpert,
S.D. Tanksley; "Cloning Transgenic Expression and Function of fw2.2:
a Quantitative Trait Locus Key to the Evolution of Tomato Fruit", Science,
accepted for publication
Please, use the folowing addresses to contact us:
Jarek Meller http://www.cs.cornell.edu/home/meller mailto:meller@phys.uni.torun.pl mailto:meller@cs.cornell.edu Ron Elber mailto:ron@cs.cornell.edu |