Research projects, collaborations, results etc ...
Structural determinants of Norwalk-like viruses specific binding to histo-blood group antigens.
Joint work with {Jason) Xi Jiang and his group.
Norwalk-like viruses (NLVs) are single-stranded, positive-sense RNA viruses that are the most important cause of non-bacterial epidemics of acute gastroenteritis, affecting individuals of all ages. NLVs appear to be recognizing, in strain specific patterns, different antigen receptors determined by blood types. Individuals who express the right types of blood group antigens become infected by a given strain while individuals who lack this strain specific receptors remain uninfected. Inspired by Jason Jiang, and in collaboration with his group, we embarked on studies that attempt to elucidate the structural determinants for the strain specific binding to different antigen receptors.
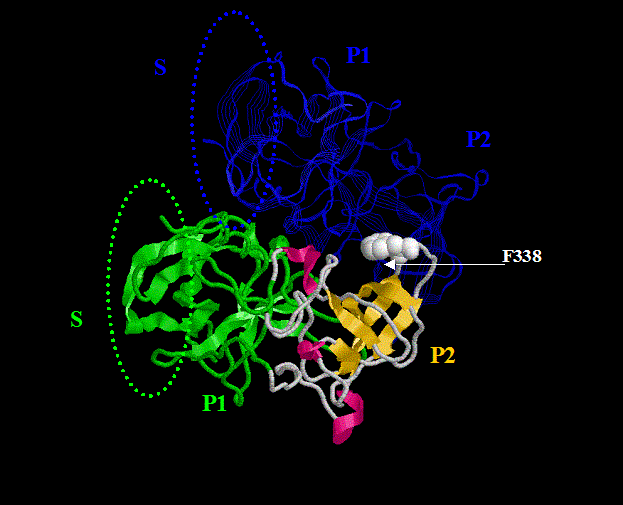
Since the prototype structure of the Norwalk virus capsid protein has been solved, we were able to use homology modeling to build models for different strains and superimpose them with evolutionary profiles for a large family of NLVs with different binding patterns. Based on such a computational analysis, a plausible binding pocket on the surface of the P2 domain (see figure) of the Norwalk-like viruses capsid proteins was identified and subsequently confirmed by mutagenesis experiments. These studies may lead to the development of drug candidates that would inhibit the binding and infection. We are planning to initiate a project to specifically design such molecules in the sprit of the rational drug design. Keep your fingers crossed!
M. Tan, P. Huang, J. Meller, W. Zhong, T. Farkas and X. Jiang; Mutations within P2 Domain of Norovirus Capsid Affect Binding to Human Histo-Blood Group Antigens: Evidence for a Binding Pocket, Journal of Virology, 77 (23): 12562-71 (2003)
Post-translational regulation of transcription: studies on protein-protein interaction in the RNA polymerase II elongation complex.
Joint work with Maria Czyzyk-Krzeska and her group.
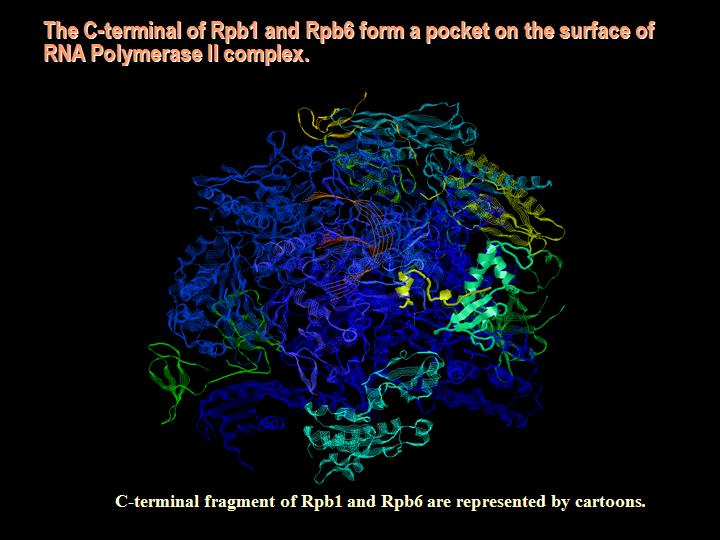
RNA Polymerase II is the major transcription complex in eukaryotes, which interacts with many co-factors in larger transcription initiation and transcription elongations complexes (with transformation between the two stages being dependent among other things on post-translation modifications, e.g., phosphorylation of the C-terminal domain of its largest subunit Rpb1, see the figure to the left and an see additional figure). Experimental hints (Maria's work) pointed out to a possible correlation between the levels of the tumor suppressor von Hippel-Lindau protein (VHL) that targets Hypoxia-Inducible Factor (HIF) for degradation under normoxic conditions, and the transcription arrest for specific genes at least. That led to the hypothesis of another factor that could be VHL-dependent and interact with RNA Pol II. either directly or indicrectly.
While searching databases for possible candidates we were able to identify Rpb1 and Rpb6 units of RNA Pol II as possibly interacting directly with the VHL protein. Using sequence-to-structure matching (threading - in order to see alignments please click here), we predicted computationally that Rpb1 (and Rpb6) is likely to share sequence and structure similarity with the Oxygen Dependent Domain (ODD) of HIF1a. Thus, VHL would be likely to interact directly with Rpb1/Rpb6 complex, as it interacts with the ODD domain of HIF1a. The critical proline residue is included in the binding site on the Rpb1 counterpart of the ODD domain of the HIF - see additional figure.
This interaction was subsequently confirmed experimentally by Maria and her group. The interaction between RNA Pol II and VHL leads to ubiquitinatiion of the largest subunit of RNA Pol II and has a potential impact on the stability of the transcription elongation complex. We are now involved in further studies on the VHL protein and its interactions.
A. V. Kuznetsova, J. Meller, P. O. Schnell, J. A. Nash, Y. Sanchez, J. W. Conaway, R. C. Conaway and M. F. Czyzyk-Krzeska; VHL binds hyperphosphorylated large subunit of RNA Polymerase II through a proline hydroxylation motif and targets it for ubiquitination, PNAS vol. 100 (5), 2706-2711 (2003)
M. F. Czyzyk-Krzeska and J. Meller; Von Hippel-Lindau Tumor Suppressor: Not Only HIF’s Executioner, Trends in Molecular Medicine, to appear (2004)
Design of scoring functions for protein folding and protein threading.
The protein folding problem consists of predicting the three-dimensional structure of a protein from its amino acid sequence and remains one of the grandest unresolved challenges in computational biology and physical chemistry. In order to characterize the existing computational approaches to this problem one may distinguish two underlying principles. The so-called ab-initio protein folding simulations attempt to reproduce the actual physical folding process, with the unique three-dimensional structure of a protein postulated to correspond to a global minimum of the free energy function. The protein recognition approach, in turn, relies on the fact that a large number of protein folds are already determined. Given an appropriate scoring function, which can be thought of as a simplified folding potential, the fold recognition methods find the ``best'' template from the library of known folds, vastly simplifying the search for the native conformation, as compared to the ab-initio approach (obviously, a novel fold cannot be assigned by protein recognition approach).
The scoring functions for protein recognition can be based either on amino acid sequence similarity (as in BLAST, for example) or on measures of sequence-to-structure fitness. The latter approach (so-called threading) allows one, in principle, to find distantly homologous proteins that share the same fold without detectable sequence similarity. In practice, as indicated in the previous section, the level of success of threading is limited by the accuracy of scoring functions and the quality of alignment of a sequence into a three-dimensional structure. Therefore, most of the fold recognition methods “blend” the sequence-to-structure fitness with other “signals”, such as sequence similarity and predicted secondary structures.
We proposed a novel position specific weighting of sequence and threading scoring functions, which may be regarded as an extension of such an approach and has a potential to achieve even greater level of success, due to a larger number of degrees of freedom in the model. We also work on new methods for predicting secondary structures that draw from the most successful approaches to secondary structure prediction, such as Psi-Pred achieving up to 80% accuracy in assigning alpha, beta and coil states to amino acid sites. However, in light of the fact that critical tertiary contacts may dramatically influence local propensities for secondary structures, one cannot expect much progress here without taking into account long-range interactions (contacts) and the new method has been designed with the idea to incorporate such effects.
Providing accurate annotations and high quality alignments is of importance for rapid screening of genes that are good candidates for further experimental studies, for protein modeling and drug design and for comparative genomic, with potential to offer a better understanding of genes implicated in various diseases. Recognizing the conceptual and practical importance of protein folding and protein recognition, the community of researchers active in this field established the Critical Assessment Of Protein Structure Prediction (CASP) competitions in blind prediction of protein structures, as new standards of verification of scientific and practical merits of new methods for protein folding in silico. When our new developments are ready they will be tested during CASP.
Related goals:
Developing next version of LOOPP: a program program for protein structure prediction and design of scoring functions for protein folding and protein threading. Launching the Sequence Independent Filtering Tool (SIFT) server for filtering and enhancement of "ab initio" folding simulations.
R. Adamczak, A. Porollo and J. Meller; Accurate Prediction of Solvent Accessibility Using Neural Networks Based Regression, Proteins: Structure, Function and Bioinformatics, to appear (2004)
A. Porollo, R. Adamczak, M. Wagner and J. Meller; Maximum Feasibility Approach for Consensus Classifiers: Applications to Protein Structure Prediction, Proceedings of The Second International Conference on Computational Intelligence, Robotics and Autonomous Systems, CIRAS 2003
R. Adamczak and J. Meller; On the Transferability of Folding and Threading Potentials and Sequence-Independent Filters for Protein Folding Simulations, submitted
A. Porollo, R. Adamczak and J. Meller; Polyview: A Flexible Visualization Tool for Structural and Functional Annotations of Proteins, Bioinformatics, to appear (2004)
Gene prediction in Pneumocystis carinii.
Joint work with Melanie Cushion and her group, within the Pneumocystis Genome Project.
The major goals of this
project are to develop new methods and tools for gene prediction in the
fungal pathogen Pneumocystis carinii (Pc) using integrated
strategies for gene prediction and discovery that couple progress in
experimental efforts in sequencing and EST discovery with computational
analysis. These objectives represent the necessary initial steps leading to a
full annotation of the Pc genome.
Organisms known as “Pneumocystis
carinii” (Pc) are non-filamentous fungi that inhabit the lungs of
mammalian hosts, where they
can exist without consequence in hosts with intact immune systems. However,
once the immune system becomes compromised, the organisms proliferate within the
lung alveoli and cause a lethal pneumonia (PcP) if untreated. No species of
Pneumocystis can be cultivated outside the mammalian lung, impeding diagnostic
capabilities as well as basic scientific research. Limited therapy is available
with which to treat the pneumonia, since these organisms are not susceptible to
standard anti-fungal drugs such as amphotericin B and fluconazole.
While gene prediction from
the primary DNA sequence remains a challenging problem, it facilitates greatly
experimental analysis, annotation and biological discovery in newly sequenced
genomes. Even though the existing methods achieved only a moderate accuracy,
computational gene prediction has become a widely used strategy in
genomic research. Standard approaches to gene
prediction are based on machine learning, pattern recognition and statistical
methods that are trained (by optimizing their adaptive parameters) to recognize
exons, introns and specific signals at splice junctions. Genome specific biases
in constitution and statistics of such signals imply that the best results are
achieved when a gene recognition system is trained on a set of examples that
represent specific characteristics of a given genome. For example, the
widely used GenScan and HMMgene programs, which employ the Hidden
Markov Model (HMM) approach, achieve about 80% accuracy (at the exon level) for
the human genome, but they had dramatically lower accuracy when applied
(in our preliminary tests) to fungal genomes without re-training their
parameters. Many other pattern recognition methods, such as neural networks and
decision trees, have been applied to the gene prediction problem as well. In
order to improve the accuracy of prediction, pattern recognition and statistical
approaches are often combined with “splicing alignments” that utilize
similarity with known genes. Such a combined strategy has been successfully
applied to the human and other genomes.
Juvenile Rheumatoid Arthritis project.
Joint work with David Glass and his group.
Progress in
developing new approaches to computational analysis and interpretation of the
growing body of genomic data, such as microarray expression profiles and
information about individual variations in disease implicated genes, is critical
for further advances of medical research. One of the main challenges for
interpreting the vast amounts of genomic information is the limitation of the
currently used algorithms and analytical tools in terms of the size and
complexity of the data. Successful translation of the basic research into
clinical practice will further require methods for integration with other
sources of information, such as data from clinical
trials.
JRA is a heterogeneous group of chronic arthropathies, which together are the most prevalent pediatric rheumatic diagnosis, affecting approximately 1-3 children in 1000. The subtypes of JRA differ in their clinical manifestation and the underlying immunogenetic characteristics, involving primarily the major histocompatibility complex (MHC). For example, the susceptibility to pauciarticular vs. polyarticular JRA types has been associated with certain alleles of (class I and II) genes from the human leukocyte antigen (HLA) region on chromosome 6. Severity and the outcome of JRA vary greatly, with polyarticular and systemic types evolving into severe cases more often than pauciarticular type. Better understanding of complex genetic and environmental contributions to the disease risk would greatly increase the chances for correct diagnosis and treatment, especially in severe cases of joint erosion that at present cannot be predicted in the early stages of the JRA onset.
The problem we address is the identification of complex genetic traits, gene-gene interactions and environmental factors triggering the onset of a disease, underlying different subtypes and outcomes of JRA. Clinically, the different types of JRA are classified according to the onset type, number of joints with active arthritis and systemic features. Using the availability of labeled data (i.e. patients with the type of JRA and clinical outcome assigned and with known results of HLA typing, expression profiles etc.) we use supervised machine learning methods for integrated analysis and classification of genomic and clinical data pertaining to JRA. Finally, it is worth stressing that the tools and methods, developed for JRA, will also be applicable to models and classification systems for other complex diseases influenced by genetic and environmental factors, such as asthma or diabetes.
Modification of protein-DNA interactions via phosphorylation: Molecular Dynamics studies on Replication Protein A.
Joint work with Kathleen Dixon and her group.
Well, we still have a way to go ...
Description of other projects soon ...